












Founded in 2013, MedGenome is a pioneer in innovative and path-breaking solutions for genetic testing with deep focus on scientific innovation and research.
Genetic Testing Portfolio




Presenting the virtual tour of the State-of-the-art Genetic Lab at MedGenome

- 3,50,000+ Exomes and Genomes sequenced
- First and only Lab with CAP accreditation for Whole Genome and Whole Exome Testing
- 1300+ Genetic tests across various diseases categories
- 700+ Clinical Geneticists, Genome Analysts, Bioinformatics Engineers
MedGenome Patient Stories
MedGenome Patient Stories

MedGenome in the News











Free Genetic Counseling by MedGenome Genetic Experts
More about MedGenome
MedGenome is committed to delivering precise, accurate, accessible, and affordable genomic solutions to empower every individual in the journey to manage their health.
Our focus as an industry leader is to provide global quality genetic testing solutions backed by technology, superior service, and innovation. We are South-East Asia’s largest CAP-accredited genetic testing laboratory, operating in India with the largest menu of over 1300 high-end genetic tests.
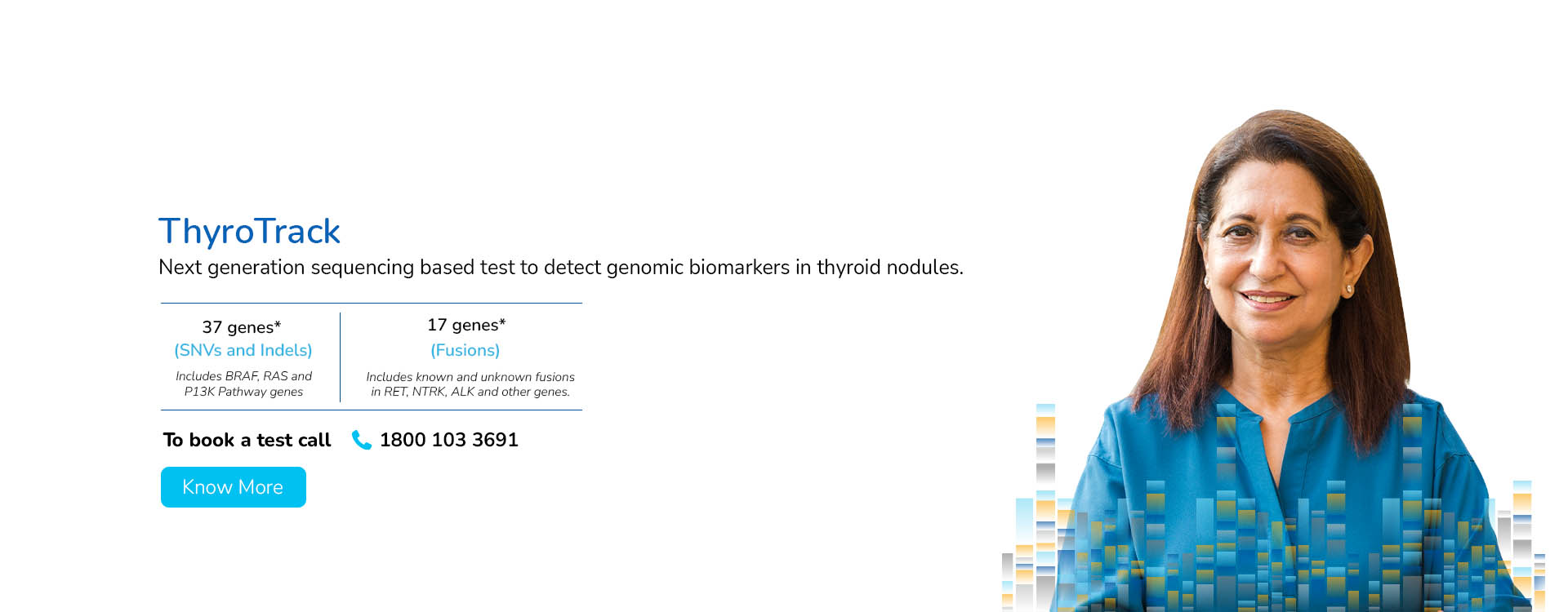
With a deep focus on scientific innovation and research, MedGenome has been leading the market with a wide range of industry-first genetic tests like Rhesus D, ThyroTrack, SPIT-SEQ, NIPT, HRR Gene Track, HRR Liquid biopsy, Whole Exome, Heme Track, Hereditary Cancer Gene Panel, Onco Track Ultima and many more for the detection and diagnosis of complex diseases.
MedGenome works closely with Clinicians to understand their challenges and expectations for effective patient treatment and happy outcomes. We follow global accreditation and quality benchmarks like CAP, EFI, PCP NDT, PCB, NABL, and many more. We are committed to delivering high-quality genetic testing solutions that are affordable and accessible to billions of people across the globe. Our proprietary tools like Varminer, Oncominer, etc. follow ACMG guidelines and provide Germline and Somatic genetic analysis with greater precision.
 Enquire
Now
Enquire
Now