Thank you for reaching out to us.




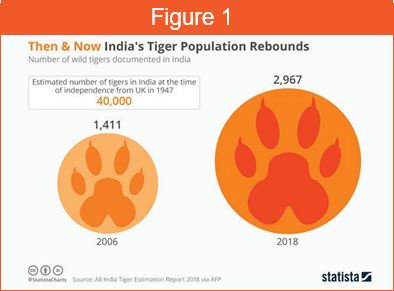
Thanks to extensive conservation efforts by various governments and NGOs, their population globally is growing back at a steady rate (Figure 1). In spite of these efforts, there is a deeper need to understand how these species have diverged and isolated in patches of forest cover across Asia. One way to achieve this is to look at their genomes.
Genome sequencing data is critical to estimate genetic relatedness in an elusive species like the tiger. Typically, studies like this need sampling the animals in the wild which may involve capturing, tagging and blood/tissue collection. Although this can be easily done with the herbivores or other small animals, for an elusive large carnivore such as the tiger, it is a challenge or rather dangerous to immobilize the animal. For such species, samples such as scat, urine, saliva, skin, hair or other environmental DNA is more feasible but is not without its own set of challenges. For example, scat samples may be dominated by microbial and prey DNA while the urine sample may be contaminated with environmental DNA. Saliva samples at the kill site may belong to more than one individual and may have prey DNA. In contrast, shed hair samples are expected to be cleaner than the above non-invasive sources.
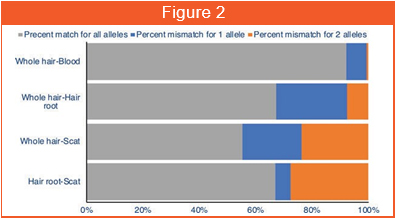
In the last couple of years, we have been working with Dr. Uma Ramakrishnan and her team at NCBS, Bangalore to generate sequencing data for different tiger populations. We have been blood, tissue, scat, hair, etc. for library preparation, sequencing and analysis. In this study, during the field work in Ranthambore Tiger Reserve, individual tigers were followed (at safe distances) and the hair samples were collected wherever the tigers rested or groomed. Later on, these samples were processed further for DNA extractions and sequencing. It turned out that DNA content from shed hair was better than that from scat samples (which was traditionally collected). It was also found that DNA sequences from shed hair were concurrent with the blood samples and yielded identical results on population connectivity (Figure 2). Using the sequences from shed hair, two new matrilines in Ranthambore Tiger Reserve were discovered. It was also discovered that the tiger T47 might belong to the same matriline as The famous tigress Machali. T47's ancestry was previously unknown. Hence, shed hair from field-observed tigers could help sequence the genetic material and establish previously unknown genetic links. The results of this work was published in the journal Ecology and Evolution (DOI: 10.1002/ece3.6157).
This study presents the largest collection of Indian tiger genomes to date including the whole genome of the tigress Machali, one of the founders of the Ranthambore tiger population (after the population recovered from a significant decline in 2005). Her genome is important as it can potentially yield insights into the ancestry of many tigers in the Ranthambore tiger reserve. The study reports whole genome sequences of six Ranthambore tigers including that of the tigers Ustad (T24), Jhumroo (T20) and Machali (T16), four of which are from shed hair and rest from blood. The key aspect to this study has been the long-term data on tiger maternity collected by the Rajasthan Forest Department. In an applied context, these results will be useful in selecting founders for tiger rewilding and translocation. From a biological perspective, this data will be critical to understanding the impacts of isolation (and connectivity) on the genetic variation of particular tiger population, and for the study of particular individual. This will in turn help in sustainable conservation of these striped cats.